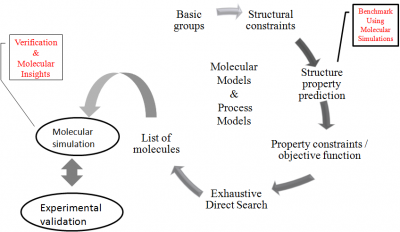
We are working on the development of computational schemes for rational solvent design to select the optimal solvent (or design a new solvent) for the extraction of a pharmaceutical intermediate, synthesised using a biotransformation process. Molecular simulations have been employed to benchmark the properties of the molecules which are estimated using the quick though inherently approximate group contribution methods in the computer aided molecular design scheme. An additional step, whereby the cost-effective molecular simulation approach (the accuracy of this approach is limited only by that of the force field employed to model the interactions in the molecule) is used to verify the trends in the computer-aided molecular design results, has also been introduced in the computational scheme. Further, the molecular simulation techniques also allow us to gain molecular insights into the solvent extraction process.
Prof. J Adhikari